NucleosomeDB main controls: searching and browsing.
Users can search for structures using either quick or advanced search buttons.

Search results page as well as collection and browse page consists of a table with a brief structure description. Queries can be additionally edited and filtered.

Search results page as well as collection and browse page consists of a table with a brief structure description. Queries can be additionally edited and filtered.
Structure information page.
The information tab holds structure provenance data and links to the source databases. 3D preview tab provides interactive visualization of the nucleosome structure in a unified color scheme, independent from PDB model chain identifiers. Every structure is aligned to a nucleosome reference frame (see below) and available for download. Additionally, the information tab contains a list of nucleosome cores and histone chains detected in the structure.

Structure analysis tools.
The nucleosome reference frame (NRF) is determined by the superhelical axis of nucleosomal DNA (Z-axis), the pseudo symmetry dyad axis (Y-axis), and the vector product of Y-axis and Z-axis (X-axis).

The core geometry tab provides a visual representation of nucleosome core atom coordinates within NRF, which is useful for previewing overall nucleosome geometry and deformations.

The DNA geometry tab provides interactive DNA structural information via relative twist plots (see Methods) and local base step parameters as defined in 3DNA software.

The DNA contacts tab provides interactive profiles of DNA-protein contacts with histone and non-histone proteins. Additional information is provided for every residue on mouse hover.

The core geometry tab provides a visual representation of nucleosome core atom coordinates within NRF, which is useful for previewing overall nucleosome geometry and deformations.
The DNA geometry tab provides interactive DNA structural information via relative twist plots (see Methods) and local base step parameters as defined in 3DNA software.
The DNA contacts tab provides interactive profiles of DNA-protein contacts with histone and non-histone proteins. Additional information is provided for every residue on mouse hover.
Example of structure comparison.
The structure of bound to OCT4-SOX2 (6YOV) is a good example of drastic nucleosome DNA conformational changes.

Add structures 6YOV and 6T93 to the collection and click the "Compare DNA parameters" button. The graphs show that DNA significantly changes the conformation in the region of the +60 position relative to the dyad axis. The relative twist graph shows that the DNA is shifted by one basepair relative to the free structure. Similarly, a conformational change is visible in the Roll parameter graph.

DNA-histone contact profile is also significantly different and clearly shows the binding region.

Add structures 6YOV and 6T93 to the collection and click the "Compare DNA parameters" button. The graphs show that DNA significantly changes the conformation in the region of the +60 position relative to the dyad axis. The relative twist graph shows that the DNA is shifted by one basepair relative to the free structure. Similarly, a conformational change is visible in the Roll parameter graph.
DNA-histone contact profile is also significantly different and clearly shows the binding region.
Nucleosome structure
Nucleosomes are fundamental blocks of chromatin compaction, which organize ~200 DNA base pairs using an octamer of histone proteins. A complete nucleosome consists of four types of core histones (H3, H4, H2A, and H2B — two copies of each) [1,2].Alterations to histone or DNA sequences, binding to chromatin proteins modulate nucleosome structure and dynamics thus provide epigenetic regulation to the DNA processing pathways (transcription, replication, repair, etc.) [3].
In recent years the number of new nucleosome structures in various conformational states had risen significantly. Such an increase is driven by the Cryo-EM revolution as it allows us to determine the structure of nucleosome complexes with various chromatin proteins. Such growth however is shadowed by the variations in used naming nomenclatures, chain identificators, and especially amino acids and nucleobases numbering. In our database, we made an effort to bring all structures to a single form.
Database scope
This database aims to be a full collection of all published nucleosome structures. Database key features are:- The database contains structures of various chromatin proteins that interact with nucleosomes.
- Histone sequences are classified using HistoneDB 2.0 nomenclature and sequences, DNA sequences were classified using this sequence set.
- All structures are analyzed, and structurally aligned for researcher convenience (see Methods), DNA residues numbers are also mapped to the nucleosome dyad axis for convenient DNA structure analysis.
- The database includes a set of analysis instruments designed for histone and DNA structure analysis as well as protein DNA contact analysis.
References
- Kornberg RD. "Chromatin structure: a repeating unit of histones and DNA." Science, 1974. PMID: 4825889
- Luger K, Mader AW, et al. "Crystal structure of the nucleosome core particle at 2.8 A resolution." Nature, 1997. PMID: 9305837
- Armeev GA, Gribkova AK, et al. "Linking chromatin composition and structural dynamics at the nucleosome level." Curr Opin Struct Biol, 2019. PMID: 30529788
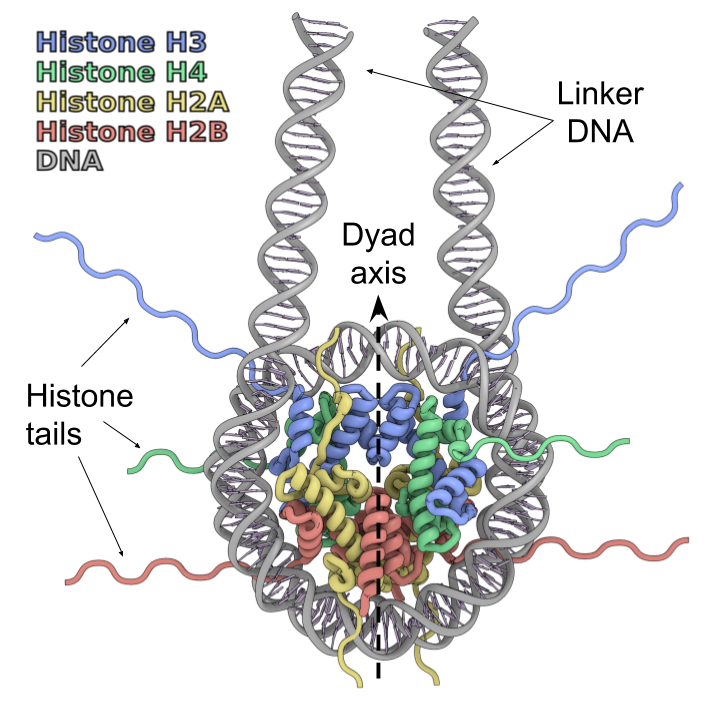 Nucleosome organization
Nucleosome organization
Data aquisition and classification
All structures were extracted from RCSB using BLAST search against histone variant sequences.Resulting hits were filtered and only structures containing DNA and all four core histone types (H3, H4, H2A, and H2B) were selected.
Histone sequences were classified using HistoneDB 2.0 nomenclature and sequences, DNA sequences were classified using DNA sequence set, extracted from literature. Chain IDs were assigned to proximal and distal halves of nucleosomes based on the contacts between histones and DNA
All structures, which contain single nucleosome and continuous DNA chains (without base modifications) were analyzed with a pipeline of instruments for nucleosome geometry and contact analysis which contains several steps.
Structure allignment to Nucleosome Reference Frame
All structures were aligned to the reference structure (1KX5) using the structural alignment of histone secondary structure elements. Initially, a reference structure was put to the nucleosomal superhelical reference frame [1]. In brief: the nucleosome reference frame is determined by the superhelical axis of nucleosomal DNA (Z-axis), the pseudo symmetry dyad axis (Y-axis), and the vector product of the Y-axis and Z-axis (X-axis).
DNA dyad detection and residue renumbering
Dyad position was assigned after structure alignment. Nucleic bases on top and bottom DNA strands were chacked with geometrical criterion: the closest to the Y-axis nucleic base was assumed to be on dyad. DNA residues numbers were then mapped to position relative to dyad which allows users to compare DNA characteristics uniformly.Analysis tools
Histone geometry analysisThis tool is designed to view and compare key nucleosome structure elements (alpha-2 helix and DNA) in projections of the nucleosome reference frame. Calpha atoms are shown for histones and centers of base pairs are calculated with 3DNA.[2]
Relative twist and local geometry analysis of DNA
A relative twist is a metric for the rotational periodicity of DNA in a nucleosome. It is defined for each base pair as the angle between the base pair vector (vector between C2' atoms of juxtaposed bases in the base pair) and the nucleosomal superhelical axis. Maxima and minima of relative twist values correspond to the integer and half-integer superhelical locations values. Local base step parameters were calculated using 3DNA [2] software. Base pairs were assigned by the complementarity of top and bottom DNA strands, as 3DNA find_pair utility can skip base pairs if DNA is deformed. Different options of sequence highlighting are available to mark stiff poly-A tracts and flexible TA and YR (pyrimidine-purine) steps.
DNA-protein contact analysis
When searching for contacts, a cut-off of 3.9A is used. Two residues are noted as contacting if at least a pair of their atoms are closer than the cutoff distance.
Interactors classification
A functional classification for chromatin proteins was designed based on [3,4,5,6,7,8,9], Gene Ontology. Nucleosomal interactors were mapped to this classification by reit Uniprot IDs and HGNC Symbols.
References
- Shaytan AK, Armeev GA, et.al. Coupling between Histone Conformations and DNA Geometry in Nucleosomes on a Microsecond Timescale: Atomistic Insights into Nucleosome Functions. J Mol Biol. PMID:26699921
- Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003. PMID:12930962
- Musselman CA, Lalonde ME, Côté J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012. PMID: 9305837
- Xu Y, Zhang S, Lin S, et al. WERAM: a database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. 2017. PMID: 27789692
- Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol. 2013. PMID: 23288364
- Mani U, S AS, Goutham R N A, Mohan S S. SWI/SNF Infobase-An exclusive information portal for SWI/SNF remodeling complex subunits. PLoS One. 2017. PMID: 28961249
- Zhang P, Torres K, Liu X, Liu CG, Pollock RE. An Overview of Chromatin-Regulating Proteins in Cells. Curr Protein Pept Sci. 2016. PMID: 26796306
- Khare SP, Habib F, Sharma R, Gadewal N, Gupta S, Galande S. HIstome - a relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic Acids Res. 2012. PMID: 22140112
- Han H, Cho JW, Lee S, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018. PMID: 29087512
- Mayran A, Drouin J. Pioneer transcription factors shape the epigenetic landscape. J Biol Chem. 2018. PMID: 29507097
...
Download
All alligned structures can be downloaded individually.To download all alligned nucleosomes structures please use this link
DNA Variants
Even though nucleosomes are abundant in chromatin and are placed relatively regularly on DNA strands, they are known to prefer some sequence patterns to others. The variability of available sequences in vivo is virtually unlimited but in vitro only a handful of sequence patterns is used as they form stable and structurally homogeneous nucleosomes.Nucleosome positioning DNA sequences were extracted from listed papers and can be obtained here here
| Name | Length | Dyad position(s) | doi |
|---|---|---|---|
| 601 | 232 | 133 | 10.1080/073911010010524947 |
| 601L | 146 | 73 | 10.1093/nar/gks261 |
| 601_modified | 146 | 73 | 10.1126/science.aau9904 |
| MMTV | 438 | 114,311 | 10.1080/073911010010524947 |
| alphasat_147 | 147 | 73 | 10.1016/S0022-2836(02)00386-8 |
| alphasat_146 | 146 | 73 | 10.1016/S0022-2836(02)00386-8 |
| alphasat_146_4CpG | 146 | 73 | 10.1002/2211-5463.12064 |
| alphasat_146b | 147 | 73 | 10.1016/S0022-2836(02)00386-8 |
| alphasat_native | 145 | 72 | 10.1038/s41467-019-10247-4 |
| alphasat_A16 | 146 | 73 | 10.1016/j.jmb.2004.07.080 |
| alphasat_4_palindorm | 147 | 73 | 10.1038/nature10258 |
| alphasat_2L | 145 | 73 | 10.1098/rsob.150128 |
| alphasat_2R | 146 | 73 | 10.1098/rsob.150128 |
| DNA-1 | 148 | 74 | 10.1038/s41586-020-2195-y |
| Human-D02 | 146 | 73 | 10.1038/s41467-019-12007-w |
| telomeric_human | 146 | 73 | 10.1101/2019.12.18.881755 |
G.A. Armeev, A.K. Gribkova, and A.K. Shaytan, Integrative Biology Group